关于DMF文件的介绍
前言
笔者在查资料的过程中发现,欧洲的ASMFs最初的讨论文件是在2006年,CEPs最开始的起源,也得到2001年(EDQM是根据2001/83 EC指令来进行CEPs认证的)。而美国的DMF指南,1989年就开始实施了,并且一直延续到现在。说起活性物质主文件,美国DMF制度是首创,欧洲以及其他后续的加拿大、澳大利亚等,都是在仿美国的DMF制度。
DMF制度确实是一个创新,对活性物质、辅料、药包材的生产商(DMF的持有者)、FDA和制剂上市申请者都有好处:一是活性物质或辅料或药包材的生产商只需要向FDA递交资料,不需要把资料给制剂上市申请者,这样保护了活性物质或辅料或药包材的生产商的商业机密。二是不同制剂上市申请者用到相同的DMF,FDA也不需要多次审评,节约了审评资源。三是对制剂上市申请者来说可以把主要精力放在制剂上。所以介绍活性物质的主文件,不得不介绍美国DMF制度。
DMF简介
一套DMF(Drug Master File)文件是递交给FDA的,包含在生产、操作、包装和储存一个或多个人用药过程中,使用到的厂房,操作流程或使用的物质的保密细节信息。DMF不是法律法规要求递交的,而是企业决定要不要递交。DMF包含的内容可支持: 临床研究申请(Investigational New Drug, IND), 新药注册(New Drug Application,NDA) 和仿制药注册(Abbreviated New Drug Application, ANDA), 另一个DMF,或是出口申请注册(Export Application),以及这些申请注册的变更或是补充申请。
DMF注册
DMF有五种类型,其中:2000年后,I型DMF FDA已不再接受。
I 型:组织与人员、设施与设备和标准操作程序;
II型:原料药、中间产品及其原料,制剂药;
III型:包装材料——容器;
IV型:赋形剂,着色剂,香料及其原料;
V型:可被FDA接受的其它信息;
DMF的状态分成两种,
“A”=active:激活状态,意味着DMF可用,没有关闭;
“I”=Inactive:未激活状态,意味着DMF被DMF持有者或FDA关闭。
重新激活DMF
未激活的DMF(或是关闭的DMF)想变成激活状态,只能通过递交激活申请来激活。激活申请应包含一整套DMF文件,并符合现行法规。封面信必须注明是激活申请。或者DMF持有者可以递交一个新的DMF。
FDA每个季度对DMF的列表清单进行更新,可在官网找到Excel版和TXT版的列表。目前DMF清单是2016年6月30日之前收到的文件,确认接受信函(acknowledgment letters)在2016年7月6号之前发出。
笔者对这份列表进行了统计分析,截止2016年第二季度,总有29414份DMFs,其中52%属于未激活状态。

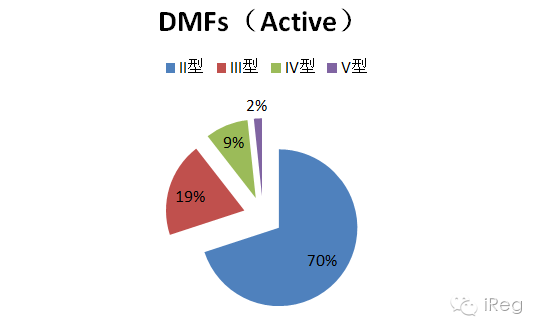
注:被取消的DMF号,预分配的DMF号和待定或是被分配到其他中的DMF号不在DMF清单中。DMF状态不传达其他信息,例如DMF是否技术审评过,或者是不是正在进行完整性审评(Completeness Assessment); 如想询问DMF是不是在完整性审评中,可发送邮件至DMFOGD@fda.hhs.gov, 进行询问。 在激活的DMF中,II型DMF最多,占有70%;V型最少,占2%;
DMF的注册格式
虽然目前为止,DMF没有要求用电子版,但到2017年5月份后,新递交的DMF都要用eCTD格式递交;如果不用eCTD格式递交,将会被CDER拒收。之前一直都用纸质递交的DMF,没有强制要求使用eCTD格式。
电子版的DMF可通过FDA的ESG递交(Eelectronic Submission Gateway). 10GB以下的注册资料都可通过ESG递交,大部分DMF都不超过10GB;FDA鼓励DMF持有者尽早获得ESG的账号。对于超过10GB的注册资料,可用物理媒介递交,例如光盘等。
在2017年5月份之前,任何时候,DMF持有者均可选择从目前的纸质DMF转到eCTD格式。如果DMF持有者决定转成eCTD格式,DMF号 (DMF number)将保持不变。如果DMF号之前是4位数的,要转成eCTD格式,需在前面补充两个00,达到6位数。例如,纸质版时,DMF号是1234,转成eCTD时,DMF号位001234;此外,DMF持有者决定转成eCTD格式,如果因为格式的转变,DMF的内容需要调整变更,这些调整和变更需要在封面信(Cover Letter)上写清楚。
2017年5月之前,对现行纸质版DMF的注册资料,需要两份纸质版;如果有DVD或CD和纸质版的注册资料一起递交,DVD或是CD不被接受。通过e-mail递交的注册资料也不被接受。纸质版的注册资料可以双面打印。
预分配DMF号
DMF持有者希望电子递交DMF文件必须事先获取一个DMF预分配号。纸质递交DMF的持有者也可获取。
公司如果想获得V型的DMF号,应获得DMF的清晰理由(Clearance);如果没有获得Clearance,在申请获得DMF号时,应在说明清楚DMF所包含的内容,例如无菌处理设施申请中CMC(Chemistry, Manufacturing and Controls)部分的资料编写格式。
DMFs应该根据CTD格式来编写。MaPP(Manual of Policies and Procedures) 中包含了审评者的职责。审评者按照Question-based Review(QbR) format 来审评。所有II型DMF中的原料药和制剂部分,也可参考QbR格式来写,尽管这并不是要求。
转成CTD格式
公司可能把现行的纸质的、非CTD格式的文件转成纸质的CTD格式。在这种情况下,建议DMF持有者递交补充申请,包含所有CTD格式中的section,这些内容在之前的非CTD格式中也都包含。每个部分的信息都应该是完整的和最新的。对于原料药(drug substance)和辅料需要递交3.2.S和2.3.S模块的资料。对于制剂,需要递交所有3.2.P和2.3.P模块资料。如因格式改变引起了DMF技术文件的变化,例如增加新的信息,新格式注册的封面信应说明哪些技术方面有改变。
沟通渠道
1) 所有DMFs的问题或意见可发送至dmfquestion@cder.fda.gov,除非DMFs在GDUFA下的问题的,这方面的问题可发邮件至:DMFOGD@FDA.hhs.gov
2) 所有的问题,在邮件的subject 上需要写清楚,邮件的大概内容以及与DMFs的关系.因为邮箱经常受到大量的垃圾邮件,邮件subject是空白的或无意义的文字或只有一个问号,邮件将不会被打开。
3) 如果询问某个具体的DMF,DMF号需要包含在邮件的subject中。
4) 其他关于DMF的问题,应发送到CDER-OPQ-Inquiries@fda.hhs.gov.
5) 关于询问报告的分类(reporting category)和批准了的申请中DMF有变更的问题,也应发送到上面的邮箱。
DMF审评
行政审评(Administrative Review)
FDA的文件室(document room)收到任何注册资料,包括DMF文件,都不会发出收到通知。收到资料后,原始的DMF文件进行行政审评,看看资料形式上是否是完整的。行政审评可能需要2-3周。如果DMF文件通过了行政审评,FDA会发出确认函(Acknowledgement Letter),通知DMF持有者DMF号。这之后,DMF的状态是激活的。如果没有通过行政审评,FDA将会通知DMF持有者DMF有哪些缺陷,这些缺陷修正后,DMF的状态才能变成激活。
技术审评(Technical Review)
DMFs只有在下面情况下,才进入技术审评:
1. DMF是激活状态.
2. DMF持有者递交了一式两份授权信(Letter of Authorization, LOA)(如果是纸质版递交);如果DMF是按照CTD格式来写的,无论是电子版还是纸质版,LOA应该包含在section 1.4.1中。LOA中应包含DMF号(DMF number).
3.DMF持有者递交一份LOA给授权方(一般为制剂生产者)。
4.授权方递交注册资料给FDA,注册资料中包含复制的LOA。复制的LOA应包含在section 1.4.2中。
完整性审评(Complete Assessment)
在GDUFA (Generic Drug User Fee Action, 仿制药付费者法案 )中规定支持仿制药申请的 II型DMF才需要进行完整性审评。参见指南”Completeness Assessments for Type II API DMFs Under GDUFA”(CA Guidance);
仿制药付费者法案包括了DMF费用条款,完整性审评以及和DMF持有者的交流。GDUFA只适用于支持ANDA申请的原料药(Active Pharmaceutical Ingredients, APIs)II型DMF。GDUFA中不适用于的II型DMF,如下:
其他种II型,IV型和V型的DMF;
其他II型DMFs:
仅支持NDAs或INDs;
API中间体
APIs制备过程中的物质
制剂中间体
制剂
通过完整性审评的DMFs有个Excel表格可以查询,在FDA网站上可以找到。只有在这张表格中找到的DMFs才通过了完整性审评。目前这个excel表格每周更新一次。
GDUFA下的II型DMF通过行政性审评(Administrative Review)并且已经按照符合相关规定,将会排队等待完整性审评。完整性审评不需要LOA。完整性审评所需要的时间要看工作量,可能需要很多周。
注:审评时DMFs文件应该是最新的,根据DMFs法规(21CRF 314.420(c)):
任何增加,改变或删除DMFs的文件(除非是此法规中paragraph(d)中规定的) 都需递交一式两份资料,并写上所影响的DMF的名称、参考号(reference number)、卷号和页码信息。
DMF指南文件建议DMF持有者一年更新一次DMFs。为了确保DMFs文件是最新的,FDA发出逾期通知信函(Overdue Notification Letter, ONLs)给DMF持有者,如果DMF持有者在过去36个月都没有递交年报。如果DMF持有者在90天内,没有对FDA的逾期通知函回复,FDA将会关闭DMF。
各类型DMF相关指南
I型
从2000年后,不再接受I型DMFs。II型、III型和IV型DMFs不需要把设备、人员或是总体的操作流程包含在DMFs里面。只需要包含DMF持有者的地址、生产地址和联系人的地址信息等。
II型 原料药和原料药中间体
参考指南:
1)"Guidance for Industry M4Q: The CTD - Quality".
2)Guidance for Industry: ANDAs: Stability Testing of Drug Substances and Products: Questions and Answers DRAFT GUIDANCE
3) GDUFA
4) ICH 指南:Q11 Development and Manufacture of Drug Substances
III型 包装材料—容器
参考的指南:
1)Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Documentation
2)MAPP 5015.5 CMC Reviews of Type III DMFs for Packaging Materials
III型DMF应用CTD格式来编写。单个的部件(Single components)和构建材料(materials of construction) 应按照原料药格式来编写。例如,生产制造过程应包含在S.2里面。组装的包装系统(Assembled container closure systems)可作为制剂的格式来写,例如构建材料可写在P.1里面 (3.2.p.1: deion and composition of the drug product), 最终包装材料的放行标准可包含在P.5(3.2.P.5: control of drug product)中。
IV型:赋形剂,着色剂,香料及其原料
参考的指南:
1)DMF Guidance
2)Guidance for Industry: Nonclinical Studies for the Safety Evaluation of PharmaceuticaExcipients”(如有毒理研究的CMC信息包含在IV型的DMF中)
IV型DMF如只包含单一的物质,可用CTD中原料药的格式来写;如包含的是混合物,例如香料的混合物,可用CTD中的制剂格式来写。如果资料中包含毒理研究的CMC信息,毒性研究的CMC信息应包含在另外的一卷或几卷中。尽管最好是把独立研究的CMC信息作为V型DMF来递交。在电子递交的辅料DMF中,关于毒性研究的信息应该包含在合适的模块中。
V型:可被FDA接受的其它信息
参考的指南:
1)21 CFR 314.420(a)
2)Guidance for Industry for the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products
根据法令,DMF持有者如果想申请V型DMF,必须给FDA清晰的理由(obtain clearance from FDA)(除了上述第二个指南中的无菌处理设施)。潜在的V型DMF持有者应该将他们的请求发送到dmfquestion@cder.fda.gov,
包括下面的内容:
1. 申请V型DMF的必要性
2. 拟定的DMF名称
3. 这些信息不能包含在IND,NDA或是ANDA中的理由
免责声明:
本文信息部分内容来自网络文章,不保证所有信息、文本、图形、链接及其他项目的绝对准确性和完整性,故仅供访问者参照使用。如您(单位或个人)认为本文某部分内容有侵权嫌疑,敬请立即通知我们,我们将在第一时间予以更正或删除。